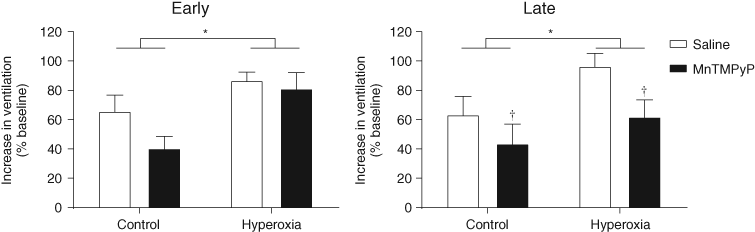
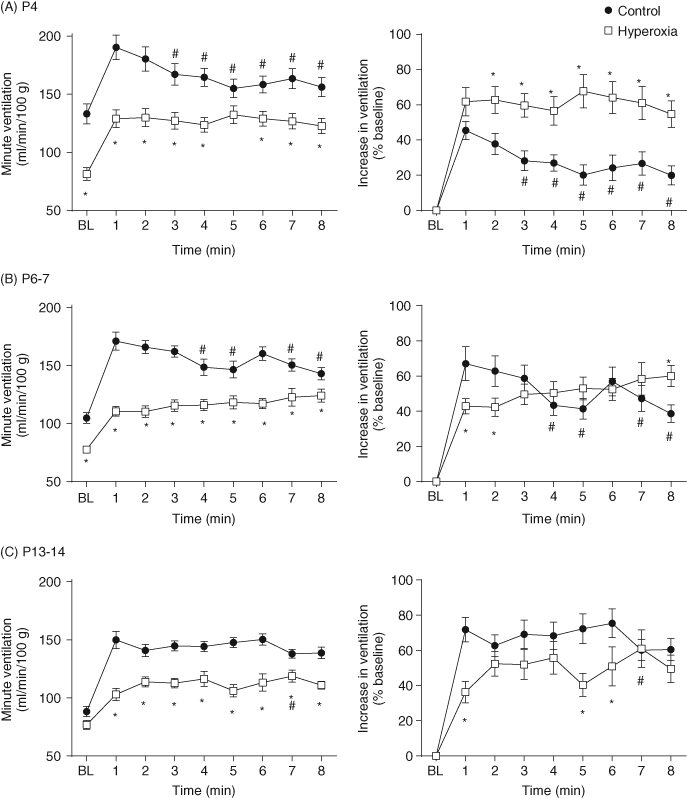
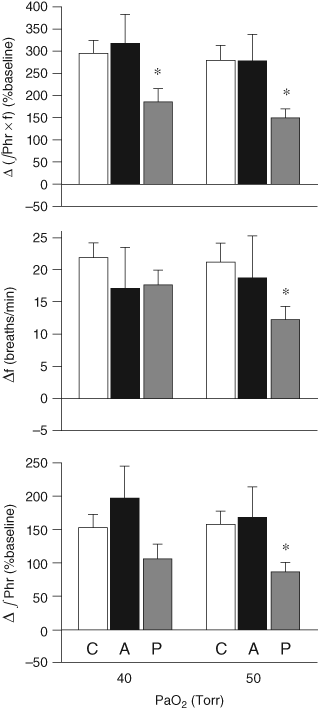
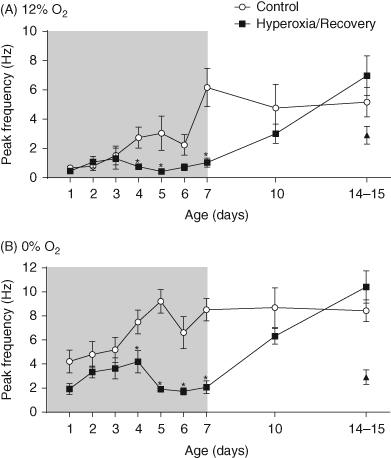
| References |
| 1. | Adolph EF, Hoy PA. Ventilation of lungs in infant and adult rats and its responses to hypoxia. J Appl Physiol 15: 1075‐1086, 1960. |
| 2. | Ainslie PN, Ogoh S, Burgess K, Celi L, McGrattan K, Peebles K, Murrell C, Subedi P, Burgess KR. Differential effects of acute hypoxia and high altitude on cerebral blood flow velocity and dynamic cerebral autoregulation: Alterations with hyperoxia. J Appl Physiol 104: 490‐498, 2008. |
| 3. | Aizad T, Bodani J, Cates D, Horvath L, Rigatto H. Effect of a single breath of 100% oxygen on respiration in neonates during sleep. J Appl Physiol 57: 1531‐1535, 1984. |
| 4. | Al‐Matary A, Kutbi I, Qurashi M, Khalil M, Alvaro R, Kwiatkowski K, Cates D, Rigatto H. Increased peripheral chemoreceptor activity may be critical in destabilizing breathing in neonates. Semin Perinatol 28: 264‐272, 2004. |
| 5. | Altman J, Bayer SA. Development of the cranial nerve ganglia and related nuclei in the rat. Adv Anat Embryol Cell Biol 74: 1‐90, 1982. |
| 6. | Alveryd A, Brody S. Cardiovascular and respiratory changes in man during oxygen breathing. Acta Physiol Scand 15: 140‐149, 1948. |
| 7. | Arieli R. Normoxic, hyperoxic, and hypoxic ventilation in rats continuously exposed for 60 h to 1 ATA O2. Aviat Space Environ Med 65: 1122‐1127, 1994. |
| 8. | Arieli R, Kerem D, Melamed Y. Hyperoxic exposure affects the ventilatory response to hypoxia in awake rats. J Appl Physiol 64: 181‐186, 1988. |
| 9. | Atanasova DY, Lazarov NE. Immunohistochemical localization of some neurotrophic factors and their receptors in the rat carotid body. Neurosci Med 4: 284‐289, 2013. |
| 10. | Atanasova DY, Lazarov NE. Expression of neurotrophic factors and their receptors in the carotid body of spontaneously hypertensive rats. Respir Physiol Neurobiol 202: 6‐15, 2014. |
| 11. | Bairam A, Niane LM, Joseph V. Role of ATP and adenosine on carotid body function during development. Respir Physiol Neurobiol 185: 57‐66, 2013. |
| 12. | Balkowiec A, Katz DM. Activity‐dependent release of endogenous brain‐derived neurotrophic factor from primary sensory neurons detected by ELISA in situ. J Neurosci 20: 7417‐7423, 2000. |
| 13. | Balkowiec A, Katz DM. Cellular mechanisms regulating activity‐dependent release of native brain‐derived neurotrophic factor from hippocampal neurons. J Neurosci 22: 10399‐10407, 2002. |
| 14. | Balkowiec A, Kunze DL, Katz DM. Brain‐derived neurotrophic factor acutely inhibits AMPA‐mediated currents in developing sensory relay neurons. J Neurosci 20: 1904‐1911, 2000. |
| 15. | Bamford OS, Carroll JL. Dynamic ventilatory responses in rats: Normal development and effects of prenatal nicotine exposure. Respir Physiol 117: 29‐40, 1999. |
| 16. | Bamford OS, Sterni LM, Wasicko MJ, Montrose MH, Carroll JL. Postnatal maturation of carotid body and type I cell chemoreception in the rat. Am J Physiol 276: L875‐L884, 1999. |
| 17. | Bancalari E, Claure N. Control of oxygenation during mechanical ventilation in the premature infant. Clin Perinatol 39: 563‐572, 2012. |
| 18. | Basting TM, Abe C, Viar KE, Stornetta RL, Guyenet PG. Is plasticity within the retrotrapezoid nucleus responsible for the recovery of the PCO2 set‐point after carotid body denervation in rats? J Physiol 594: 3371‐3390, 2016. |
| 19. | Bates ML, Farrell ET, Eldridge MW. Abnormal ventilatory responses in adults born prematurely. N Engl J Med 370: 584‐585, 2014. |
| 20. | Bates ML, Pillers DA, Palta M, Farrell ET, Eldridge MW. Ventilatory control in infants, children, and adults with bronchopulmonary dysplasia. Respir Physiol Neurobiol 189: 329‐337, 2013. |
| 21. | Bates ML, Welch BT, Randall JT, Petersen‐Jones HG, Limberg JK. Carotid body size measured by computed tomographic angiography in individuals born prematurely. Respir Physiol Neurobiol 258: 47‐52, 2018. |
| 22. | Bavis RW. Developmental plasticity of the hypoxic ventilatory response after perinatal hyperoxia and hypoxia. Respir Physiol Neurobiol 149: 287‐299, 2005. |
| 23. | Bavis RW, Blegen HJ, Logan S, Fallon SC, McDonough AB. Role of TrkB during the postnatal development of the rat carotid body. Respir Physiol Neurobiol 219: 18‐24, 2015. |
| 24. | Bavis RW, DeAngelis KJ, Horowitz TC, Reedich LM, March RJ. Hyperoxia‐induced developmental plasticity of the hypoxic ventilatory response in neonatal rats: Contributions of glutamate‐dependent and PDGF‐dependent mechanisms. Respir Physiol Neurobiol 191: 84‐94, 2014. |
| 25. | Bavis RW, Dmitrieff EF, Young KM, Piro SE. Hypoxic ventilatory response of adult rats and mice after developmental hyperoxia. Respir Physiol Neurobiol 177: 342‐346, 2011. |
| 26. | Bavis RW, Fallon SC, Dmitrieff EF. Chronic hyperoxia and the development of the carotid body. Respir Physiol Neurobiol 185: 94‐104, 2013. |
| 27. | Bavis RW, Kim I, Pradhan N, Nawreen N, Dmitrieff EF, Carroll JL, Donnelly DF. Recovery of carotid body O2 sensitivity following chronic postnatal hyperoxia in rats. Respir Physiol Neurobiol 177: 47‐55, 2011. |
| 28. | Bavis RW, Li KY, DeAngelis KJ, March RJ, Wallace JA, Logan S, Putnam RW. Ventilatory and chemoreceptor responses to hypercapnia in neonatal rats chronically exposed to moderate hyperoxia. Respir Physiol Neurobiol 237: 22‐34, 2017. |
| 29. | Bavis RW, MacFarlane PM. Developmental plasticity in the neural control of breathing. Exp Neurol 287: 176‐191, 2017. |
| 30. | Bavis RW, Millstrom AH, Kim SM, MacDonald CA, O'Toole CA, Asklof K, McDonough AB. Combined effects of intermittent hyperoxia and intermittent hypercapnic hypoxia on respiratory control in neonatal rats. Respir Physiol Neurobiol 260: 70‐81, 2019. |
| 31. | Bavis RW, Mitchell GS. Long‐term effects of the perinatal environment on respiratory control. J Appl Physiol 104: 1220‐1229, 2008. |
| 32. | Bavis RW, Olson EB Jr, Mitchell GS. Critical developmental period for hyperoxia‐induced blunting of hypoxic phrenic responses in rats. J Appl Physiol 92: 1013‐1018, 2002. |
| 33. | Bavis RW, Olson EB Jr, Vidruk EH, Bisgard GE, Mitchell GS. Level and duration of developmental hyperoxia influence impairment of hypoxic phrenic responses in rats. J Appl Physiol 95: 1550‐1559, 2003. |
| 34. | Bavis RW, Russell KE, Simons JC, Otis JP. Hypoxic ventilatory responses in rats after hypercapnic hyperoxia and intermittent hyperoxia. Respir Physiol Neurobiol 155: 193‐202, 2007. |
| 35. | Bavis RW, Simons JC. Developmental hyperoxia attenuates the hypoxic ventilatory response in Japanese quail (Coturnix japonica). Respir Physiol Neurobiol 164: 411‐418, 2008. |
| 36. | Bavis RW, van Heerden ES, Brackett DG, Harmeling LH, Johnson SM, Blegen HJ, Logan S, Nguyen GN, Fallon SC. Postnatal development of eupneic ventilation and metabolism in rats chronically exposed to moderate hyperoxia. Respir Physiol Neurobiol 198: 1‐12, 2014. |
| 37. | Bavis RW, Wenninger JM, Miller BM, Dmitrieff EF, Olson EB Jr, Mitchell GS, Bisgard GE. Respiratory plasticity after perinatal hyperoxia is not prevented by antioxidant supplementation. Respir Physiol Neurobiol 160: 301‐312, 2008. |
| 38. | Bavis RW, Young KM, Barry KJ, Boller MR, Kim E, Klein PM, Ovrutsky AR, Rampersad DA. Chronic hyperoxia alters the early and late phases of the hypoxic ventilatory response in neonatal rats. J Appl Physiol 109: 796‐803, 2010. |
| 39. | Bean JW, Rottschafer G. Reflexogenic and central structures in oxygen poisoning. J Physiol 94: 294‐306, 1938. |
| 40. | Becker H, Polo O, McNamara SG, Berthon‐Jones M, Sullivan CE. Ventilatory response to isocapnic hyperoxia. J Appl Physiol 78: 696‐701, 1995. |
| 41. | Becker HF, Polo O, McNamara SG, Berthon‐Jones M, Sullivan CE. Effect of different levels of hyperoxia on breathing in healthy subjects. J Appl Physiol 81: 1683‐1690, 1996. |
| 42. | Bentsen MH, Markestad T, Oymar K, Halvorsen T. Lung function at term in extremely preterm‐born infants: A regional prospective cohort study. BMJ Open 7: e016868, 2017. |
| 43. | Berger J, Bhandari V. Animal models of bronchopulmonary dysplasia. The term mouse models. Am J Physiol Lung Cell Mol Physiol 307: L936‐L947, 2014. |
| 44. | Berkelhamer SK, Farrow KN. Developmental regulation of antioxidant enzymes and their impact on neonatal lung disease. Antioxid Redox Signal 21: 1837‐1848, 2014. |
| 45. | Berner RA, Vandenbrooks JM, Ward PD. Evolution. Oxygen and evolution. Science 316: 557‐558, 2007. |
| 46. | Bierman AM, Tankersley CG, Wilson CG, Chavez‐Valdez R, Gauda EB. Perinatal hyperoxic exposure reconfigures the central respiratory network contributing to intolerance to anoxia in newborn rat pups. J Appl Physiol 116: 47‐53, 2014. |
| 47. | Bisgard GE, Olson EB Jr, Bavis RW, Wenninger J, Nordheim EV, Mitchell GS. Carotid chemoafferent plasticity in adult rats following developmental hyperoxia. Respir Physiol Neurobiol 145: 3‐11, 2005. |
| 48. | Bisgard GE, Olson EB Jr, Wang ZY, Bavis RW, Fuller DD, Mitchell GS. Adult carotid chemoafferent responses to hypoxia after 1, 2, and 4 wk of postnatal hyperoxia. J Appl Physiol 95: 946‐952, 2003. |
| 49. | Bissonnette JM. Mechanisms regulating hypoxic respiratory depression during fetal and postnatal life. Am J Physiol Regul Integr Comp Physiol 278: R1391‐R1400, 2000. |
| 50. | Blanco CE, Chen V, Maertzdorf W, Bamford OS, Hanson M. Effect of hyperoxia (PaO2 50‐90 mmHg) on fetal breathing movements in the unanaesthetized fetal sheep. J Dev Physiol 14: 235‐241, 1990. |
| 51. | Blanco CE, Dawes GS, Hanson MA, McCooke HB. The response to hypoxia of arterial chemoreceptors in fetal sheep and new‐born lambs. J Physiol 351: 25‐37, 1984. |
| 52. | Blanco CE, Hanson MA, McCooke HB. Effects on carotid chemoreceptor resetting of pulmonary ventilation in the fetal lamb in utero. J Dev Physiol 10: 167‐174, 1988. |
| 53. | Bouferrache B, Filtchev S, Leke A, Marbaix‐Li Q, Freville M, Gaultier C. The hyperoxic test in infants reinvestigated. Am J Respir Crit Care Med 161: 160‐165, 2000. |
| 54. | Brady JP, Cotton EC, Tooley WH. Chemoreflexes in the new‐born infant: Effects of 100% oxygen on heart rate and ventilation. J Physiol 172: 332‐341, 1964. |
| 55. | Brady R, Zaidi SI, Mayer C, Katz DM. BDNF is a target‐derived survival factor for arterial baroreceptor and chemoafferent primary sensory neurons. J Neurosci 19: 2131‐2142, 1999. |
| 56. | Brosenitsch TA, Katz DM. Physiological patterns of electrical stimulation can induce neuronal gene expression by activating N‐type calcium channels. J Neurosci 21: 2571‐2579, 2001. |
| 57. | Brosenitsch TA, Katz DM. Expression of Phox2 transcription factors and induction of the dopaminergic phenotype in primary sensory neurons. Mol Cell Neurosci 20: 447‐457, 2002. |
| 58. | Brosenitsch TA, Salgado‐Commissariat D, Kunze DL, Katz DM. A role for L‐type calcium channels in developmental regulation of transmitter phenotype in primary sensory neurons. J Neurosci 18: 1047‐1055, 1998. |
| 59. | Bucher JR, Roberts RJ. The development of the newborn rat lung in hyperoxia: A dose‐response study of lung growth, maturation, and changes in antioxidant enzyme activities. Pediatr Res 15: 999‐1008, 1981. |
| 60. | Buczynski BW, Maduekwe ET, O'Reilly MA. The role of hyperoxia in the pathogenesis of experimental BPD. Semin Perinatol 37: 69‐78, 2013. |
| 61. | Budzinska K, Ilasz R. Superoxide dismutase mimetic modulates hyperoxic augmentation of the diaphragmatic response to poikilocapnic hypoxia in non‐vagotomized rats. J Physiol Pharmacol 59 (Suppl 6): 163‐172, 2008. |
| 62. | Burggren WW, Reyna KS. Developmental trajectories, critical windows and phenotypic alteration during cardio‐respiratory development. Respir Physiol Neurobiol 178: 13‐21, 2011. |
| 63. | Burton MD, Kazemi H. Neurotransmitters in central respiratory control. Respir Physiol 122: 111‐121, 2000. |
| 64. | Busch MA, Bisgard GE, Mesina JE, Forster HV. The effects of unilateral carotid body excision on ventilatory control in goats. Respir Physiol 54: 353‐361, 1983. |
| 65. | Calder NA, Williams BA, Smyth J, Boon AW, Kumar P, Hanson MA. Absence of ventilatory responses to alternating breaths of mild hypoxia and air in infants who have had bronchopulmonary dysplasia: Implications for the risk of sudden infant death. Pediatr Res 35: 677‐681, 1994. |
| 66. | Carroll JL. Developmental plasticity in respiratory control. J Appl Physiol 94: 375‐389, 2003. |
| 67. | Carroll JL, Kim I. Postnatal development of carotid body glomus cell O2 sensitivity. Respir Physiol Neurobiol 149: 201‐215, 2005. |
| 68. | Castillo A, Sola A, Baquero H, Neira F, Alvis R, Deulofeut R, Critz A. Pulse oxygen saturation levels and arterial oxygen tension values in newborns receiving oxygen therapy in the neonatal intensive care unit: Is 85% to 93% an acceptable range? Pediatrics 121: 882‐889, 2008. |
| 69. | Cattarossi L, Rubini S, Macagno F. Aminophylline and increased activity of peripheral chemoreceptors in newborn infants. Arch Dis Child 69: 52‐54, 1993. |
| 70. | Cayetanot F, Larnicol N, Peyronnet J. Antenatal environmental stress and maturation of the breathing control, experimental data. Respir Physiol Neurobiol 168: 92‐100, 2009. |
| 71. | Chang AJ. Acute oxygen sensing by the carotid body: From mitochondria to plasma membrane. J Appl Physiol 123: 1335‐1343, 2017. |
| 72. | Chavez‐Valdez R, Mason A, Nunes AR, Northington FJ, Tankersley C, Ahlawat R, Johnson SM, Gauda EB. Effect of hyperoxic exposure during early development on neurotrophin expression in the carotid body and nucleus tractus solitarii. J Appl Physiol 112: 1762‐1772, 2012. |
| 73. | Ciarlone GE, Dean JB. Acute hypercapnic hyperoxia stimulates reactive species production in the caudal solitary complex of rat brain slices but does not induce oxidative stress. Am J Physiol Cell Physiol 311: C1027‐C1039, 2016. |
| 74. | Ciarlone GE, Dean JB. Normobaric hyperoxia stimulates superoxide and nitric oxide production in the caudal solitary complex of rat brain slices. Am J Physiol Cell Physiol 311: C1014‐C1026, 2016. |
| 75. | Ciarlone GE, Hinojo CM, Stavitzski NM, Dean JB. CNS function and dysfunction during exposure to hyperbaric oxygen in operational and clinical settings. Redox Biol 101159, 2019. DOI: 10.1016/j.redox.2019.10115. |
| 76. | Clark CG, Hasser EM, Kunze DL, Katz DM, Kline DD. Endogenous brain‐derived neurotrophic factor in the nucleus tractus solitarius tonically regulates synaptic and autonomic function. J Neurosci 31: 12318‐12329, 2011. |
| 77. | Claure N, Bancalari E. Automated respiratory support in newborn infants. Semin Fetal Neonatal Med 14: 35‐41, 2009. |
| 78. | Claure N, Bancalari E. Closed‐loop control of inspired oxygen in premature infants. Semin Fetal Neonatal Med 20: 198‐204, 2015. |
| 79. | Cohen‐Cory S, Kidane AH, Shirkey NJ, Marshak S. Brain‐derived neurotrophic factor and the development of structural neuronal connectivity. Dev Neurobiol 70: 271‐288, 2010. |
| 80. | Cragg PA, Drysdale DB, Hamilton JH. Ventilation in intact and glossopharyngeal nerve sectioned anaesthetized rats exposed to oxygen at high pressure. J Physiol 370: 489‐499, 1986. |
| 81. | Cragg PA, Khrisanapant W. Is the second carotid body redundant? Adv Exp Med Biol 360: 297‐299, 1994. |
| 82. | Cross KW, Oppe TE. The effect of inhalation of high and low concentrations of oxygen on the respiration of the premature infant. J Physiol 117: 38‐55, 1952. |
| 83. | Cross KW, Warner P. The effect of inhalation of high and low oxygen concentrations on the respiration of the newborn infant. J Physiol 114: 283‐295, 1951. |
| 84. | Cummings KJ, Frappell PB. Breath‐to‐breath hypercapnic response in neonatal rats: Temperature dependency of the chemoreflexes and potential implications for breathing stability. Am J Physiol Regul Integr Comp Physiol 297: R124‐R134, 2009. |
| 85. | Cunningham S, McColm JR, Wade J, Sedowofia K, McIntosh N, Fleck B. A novel model of retinopathy of prematurity simulating preterm oxygen variability in the rat. Invest Ophthalmol Vis Sci 41: 4275‐4280, 2000. |
| 86. | Darnall RA. The role of CO2 and central chemoreception in the control of breathing in the fetus and the neonate. Respir Physiol Neurobiol 173: 201‐212, 2010. |
| 87. | Dauger S, Ferkdadji L, Saumon G, Vardon G, Peuchmaur M, Gaultier C, Gallego J. Neonatal exposure to 65% oxygen durably impairs lung architecture and breathing pattern in adult mice. Chest 123: 530‐538, 2003. |
| 88. | Davi M, Sankaran K, Rigatto H. Effect of inhaling 100% O2 on ventilation and acid‐base balance in cerebrospinal fluid of neonates. Biol Neonate 38: 85‐89, 1980. |
| 89. | Davidson LM, Berkelhamer SK. Bronchopulmonary dysplasia: Chronic lung disease of infancy and long‐term pulmonary outcomes. J Clin Med 6, 2017. DOI: 10.3390/jcm6010004. |
| 90. | Dean JB, Mulkey DK, Garcia AJ III, Putnam RW, Henderson RA III. Neuronal sensitivity to hyperoxia, hypercapnia, and inert gases at hyperbaric pressures. J Appl Physiol 95: 883‐909, 2003. |
| 91. | Dean JB, Mulkey DK, Henderson RA III, Potter SJ, Putnam RW. Hyperoxia, reactive oxygen species, and hyperventilation: Oxygen sensitivity of brain stem neurons. J Appl Physiol 96: 784‐791, 2004. |
| 92. | Dean JB, Putnam RW. The caudal solitary complex is a site of central CO2 chemoreception and integration of multiple systems that regulate expired CO2. Respir Physiol Neurobiol 173: 274‐287, 2010. |
| 93. | Dejours P. Chemoreflexes in breathing. Physiol Rev 42: 335‐358, 1962. |
| 94. | Dennery PA, Di Fiore JM, Ambalavanan N, Bancalari E, Carroll JL, Claure N, Hamvas A, Hibbs AM, Indic P, Kemp J, Krahn KN, Lake D, Laposky A, Martin RJ, Natarajan A, Rand C, Schau M, Weese‐Mayer DE, Zimmet AM, Moorman JR. Pre‐Vent: The prematurity‐related ventilatory control study. Pediatr Res 85: 769‐776, 2019. |
| 95. | Di Giulio C, Di Muzio M, Sabatino G, Spoletini L, Amicarelli F, Di Ilio C, Modesti A. Effect of chronic hyperoxia on young and old rat carotid body ultrastructure. Exp Gerontol 33: 319‐329, 1998. |
| 96. | Dildy GA, Clark SL, Loucks CA. Intrapartum fetal pulse oximetry: The effects of maternal hyperoxia on fetal arterial oxygen saturation. Am J Obstet Gynecol 171: 1120‐1124, 1994. |
| 97. | Dmitrieff EF, Piro SE, Broge TA Jr, Dunmire KB, Bavis RW. Carotid body growth during chronic postnatal hyperoxia. Respir Physiol Neurobiol 180: 193‐203, 2012. |
| 98. | Dmitrieff EF, Wilson JT, Dunmire KB, Bavis RW. Chronic hyperoxia alters the expression of neurotrophic factors in the carotid body of neonatal rats. Respir Physiol Neurobiol 175: 220‐227, 2011. |
| 99. | Domm W, Misra RS, O'Reilly MA. Affect of early life oxygen exposure on proper lung development and response to respiratory viral infections. Front Med (Lausanne) 2: 55, 2015. |
| 100. | Donnelly DF. Chemoreceptor nerve excitation may not be proportional to catecholamine secretion. J Appl Physiol 81: 657‐664, 1996. |
| 101. | Donnelly DF. Development of carotid body/petrosal ganglion response to hypoxia. Respir Physiol Neurobiol 149: 191‐199, 2005. |
| 102. | Donnelly DF, Bavis RW, Kim I, Dbouk HA, Carroll JL. Time course of alterations in pre‐ and post‐synaptic chemoreceptor function during developmental hyperoxia. Respir Physiol Neurobiol 168: 189‐197, 2009. |
| 103. | Donnelly DF, Kim I, Carle C, Carroll JL. Perinatal hyperoxia for 14 days increases nerve conduction time and the acute unitary response to hypoxia of rat carotid body chemoreceptors. J Appl Physiol 99: 114‐119, 2005. |
| 104. | Doolette DJ, Mitchell SJ. Hyperbaric conditions. Compr Physiol 1: 163‐201, 2011. |
| 105. | Dripps RD, Comroe JH Jr. The effect of the inhalation of high and low oxygen concentrations on respiration, pulse rate, ballistocardiogram and arterial oxygen saturation (oximeter) of normal individuals. Am J Physiol 149: 277‐291, 1947. |
| 106. | Dylag AM, Raffay TM. Rodent models of respiratory control and respiratory system development—Clinical significance. Respir Physiol Neurobiol 268: 103249, 2019. |
| 107. | Eden GJ, Hanson MA. Effect of hyperoxia from birth on the carotid chemoreceptor and ventilatory responses of rats to acute hypoxia. J Physiol 374: 24P, 1986. (Abstract). |
| 108. | Eden GJ, Hanson MA. Effects of chronic hypoxia from birth on the ventilatory response to acute hypoxia in the newborn rat. J Physiol 392: 11‐19, 1987. |
| 109. | Eden GJ, Hanson MA. Maturation of the respiratory response to acute hypoxia in the newborn rat. J Physiol 392: 1‐9, 1987. |
| 110. | Elovitz MA, Mrinalini C. Animal models of preterm birth. Trends Endocrinol Metab 15: 479‐487, 2004. |
| 111. | Erickson JT, Mayer C, Jawa A, Ling L, Olson EB Jr, Vidruk EH, Mitchell GS, Katz DM. Chemoafferent degeneration and carotid body hypoplasia following chronic hyperoxia in newborn rats. J Physiol 509 (Pt 2): 519‐526, 1998. |
| 112. | Erickson JT, Millhorn DE. Hypoxia and electrical stimulation of the carotid sinus nerve induce Fos‐like immunoreactivity within catecholaminergic and serotoninergic neurons of the rat brainstem. J Comp Neurol 348: 161‐182, 1994. |
| 113. | Fidone SJ, Sato A. A study of chemoreceptor and baroreceptor A and C‐fibres in the cat carotid nerve. J Physiol 205: 527‐548, 1969. |
| 114. | Floyd TF, Clark JM, Gelfand R, Detre JA, Ratcliffe S, Guvakov D, Lambertsen CJ, Eckenhoff RG. Independent cerebral vasoconstrictive effects of hyperoxia and accompanying arterial hypocapnia at 1 ATA. J Appl Physiol 95: 2453‐2461, 2003. |
| 115. | Forster HV. Plasticity in the control of breathing following sensory denervation. J Appl Physiol 94: 784‐794, 2003. |
| 116. | Forster HV, Smith CA. Contributions of central and peripheral chemoreceptors to the ventilatory response to CO2/H+. J Appl Physiol 108: 989‐994, 2010. |
| 117. | Frank L. Developmental aspects of experimental pulmonary oxygen toxicity. Free Radic Biol Med 11: 463‐494, 1991. |
| 118. | Fuller DD, Bavis RW, Vidruk EH, Wang ZY, Olson EB Jr, Bisgard GE, Mitchell GS. Life‐long impairment of hypoxic phrenic responses in rats following 1 month of developmental hyperoxia. J Physiol 538: 947‐955, 2002. |
| 119. | Fuller DD, Wang ZY, Ling L, Olson EB, Bisgard GE, Mitchell GS. Induced recovery of hypoxic phrenic responses in adult rats exposed to hyperoxia for the first month of life. J Physiol 536: 917‐926, 2001. |
| 120. | Gamper N, Ooi L. Redox and nitric oxide‐mediated regulation of sensory neuron ion channel function. Antioxid Redox Signal 22: 486‐504, 2015. |
| 121. | Gao L, Gonzalez‐Rodriguez P, Ortega‐Saenz P, Lopez‐Barneo J. Redox signaling in acute oxygen sensing. Redox Biol 12: 908‐915, 2017. |
| 122. | Gao L, Ortega‐Saenz P, Lopez‐Barneo J. Acute oxygen sensing‐Role of metabolic specifications in peripheral chemoreceptor cells. Respir Physiol Neurobiol 265: 100‐111, 2019. |
| 123. | Garcia AJ III, Putnam RW, Dean JB. Hyperbaric hyperoxia and normobaric reoxygenation increase excitability and activate oxygen‐induced potentiation in CA1 hippocampal neurons. J Appl Physiol 109: 804‐819, 2010. |
| 124. | Garcia AJ III, Putnam RW, Dean JB. Hyperoxic stimulation of synchronous orthodromic activity and induction of neural plasticity does not require changes in excitatory synaptic transmission. J Appl Physiol 109: 820‐829, 2010. |
| 125. | Gasier HG, Demchenko IT, Tatro LG, Piantadosi CA. S‐Nitrosylation of GAD65 is implicated in decreased GAD activity and oxygen‐induced seizures. Neurosci Lett 653: 283‐287, 2017. |
| 126. | Gautier H. Hypoxia, hyperoxia and breathing. J Biosci 31: 185‐190, 2006. |
| 127. | Gautier H, Bonora M, Gaudy JH. Ventilatory response of the conscious or anesthetized cat to oxygen breathing. Respir Physiol 65: 181‐196, 1986. |
| 128. | Gelfand R, Lambertsen CJ, Clark JM, Hopkin E. Hypoxic ventilatory sensitivity in men is not reduced by prolonged hyperoxia (Predictive Studies V and VI). J Appl Physiol 84: 292‐302, 1998. |
| 129. | Georgopoulos D, Holtby SG, Berezanski D, Anthonisen NR. Aminophylline effects on ventilatory response to hypoxia and hyperoxia in normal adults. J Appl Physiol 67: 1150‐1156, 1989. |
| 130. | Gourine AV, Llaudet E, Thomas T, Dale N, Spyer KM. Adenosine release in nucleus tractus solitarii does not appear to mediate hypoxia‐induced respiratory depression in rats. J Physiol 544: 161‐170, 2002. |
| 131. | Gozal D. Potentiation of hypoxic ventilatory response by hyperoxia in the conscious rat: Putative role of nitric oxide. J Appl Physiol 85: 129‐132, 1998. |
| 132. | Gozal D, Arens R, Omlin KJ, Ward SL, Keens TG. Absent peripheral chemosensitivity in Prader‐Willi syndrome. J Appl Physiol 77: 2231‐2236, 1994. |
| 133. | Gozal D, Gozal E, Gozal YM, Torres JE. Nitric oxide synthase isoforms and peripheral chemoreceptor stimulation in conscious rats. Neuroreport 7: 1145‐1148, 1996. |
| 134. | Gozal D, Gozal E, Simakajornboon N. Signaling pathways of the acute hypoxic ventilatory response in the nucleus tractus solitarius. Respir Physiol 121: 209‐221, 2000. |
| 135. | Gozal D, Gozal E, Torres JE, Gozal YM, Nuckton TJ, Hornby PJ. Nitric oxide modulates ventilatory responses to hypoxia in the developing rat. Am J Respir Crit Care Med 155: 1755‐1762, 1997. |
| 136. | Gozal D, Torres JE, Gozal YM, Littwin SM. Effect of nitric oxide synthase inhibition on cardiorespiratory responses in the conscious rat. J Appl Physiol 81: 2068‐2077, 1996. |
| 137. | Hagadorn JI, Furey AM, Nghiem TH, Schmid CH, Phelps DL, Pillers DA, Cole CH. Achieved versus intended pulse oximeter saturation in infants born less than 28 weeks' gestation: The AVIOx study. Pediatrics 118: 1574‐1582, 2006. |
| 138. | Hanson MA, Eden GJ, Nijhuis JG, Moore PJ. Peripheral chemoreceptors and other oxygen sensors in the fetus and newborn. In: Lahiri S, Forster RE, Davies RO, Pack AI, editors. Chemoreceptors and Reflexes in Breathing: Cellular and Molecular Aspects. New York: Oxford University Press, 1989, p. 113‐120. |
| 139. | Haouzi P, Allioui EM, Gille JP, Bedez Y, Tousseul B, Chalon B. Stimulation of ventilation by normobaric hyperoxia in exercising dogs. Exp Physiol 85: 829‐838, 2000. |
| 140. | Harabin AL, Homer LD, Bradley ME. Pulmonary oxygen toxicity in awake dogs: Metabolic and physiological effects. J Appl Physiol 57: 1480‐1488, 1984. |
| 141. | Hellard D, Brosenitsch T, Fritzsch B, Katz DM. Cranial sensory neuron development in the absence of brain‐derived neurotrophic factor in BDNF/Bax double null mice. Dev Biol 275: 34‐43, 2004. |
| 142. | Herman JK, O'Halloran KD, Bisgard GE. Sustained moderate hyperoxia augments the acute hypoxic response in awake goats. Adv Exp Med Biol 499: 325‐330, 2001. |
| 143. | Hertz L. The glutamate‐glutamine (GABA) cycle: Importance of late postnatal development and potential reciprocal interactions between biosynthesis and degradation. Front Endocrinol 4: 59, 2013. |
| 144. | Hertzberg T, Brosenitsch T, Katz DM. Depolarizing stimuli induce high levels of dopamine synthesis in fetal rat sensory neurons. Neuroreport 7: 233‐237, 1995. |
| 145. | Hertzberg T, Fan G, Finley JC, Erickson JT, Katz DM. BDNF supports mammalian chemoafferent neurons in vitro and following peripheral target removal in vivo. Dev Biol 166: 801‐811, 1994. |
| 146. | Hertzberg T, Hellstrom S, Lagercrantz H, Pequignot JM. Development of the arterial chemoreflex and turnover of carotid body catecholamines in the newborn rat. J Physiol 425: 211‐225, 1990. |
| 147. | Hill CB, Grandgeorge SH, Bavis RW. Developmental hyperoxia alters CNS mechanisms underlying hypoxic ventilatory depression in neonatal rats. Respir Physiol Neurobiol 189: 498‐505, 2013. |
| 148. | Honda Y, Tani H, Masuda A, Kobayashi T, Nishino T, Kimura H, Masuyama S, Kuriyama T. Effect of prior O2 breathing on ventilatory response to sustained isocapnic hypoxia in adult humans. J Appl Physiol 81: 1627‐1632, 1996. |
| 149. | Hoop B, Beagle JL, Maher TJ, Kazemi H. Brainstem amino acid neurotransmitters and hypoxic ventilatory response. Respir Physiol 118: 117‐129, 1999. |
| 150. | Huang J, Suguihara C, Hehre D, Lin J, Bancalari E. Effects of GABA receptor blockage on the respiratory response to hypoxia in sedated newborn piglets. J Appl Physiol 77: 1006‐1010, 1994. |
| 151. | Huang YH, Brown AR, Cross SJ, Cruz J, Rice A, Jaiswal S, Fregosi RF. Influence of prenatal nicotine exposure on development of the ventilatory response to hypoxia and hypercapnia in neonatal rats. J Appl Physiol 109: 149‐158, 2010. |
| 152. | Iturriaga R, Alcayaga J, Zapata P. Dissociation of hypoxia‐induced chemosensory responses and catecholamine efflux in cat carotid body superfused in vitro. J Physiol 497 (Pt 2): 551‐564, 1996. |
| 153. | Iturriaga R, Andrade DC, Del Rio R. Enhanced carotid body chemosensory activity and the cardiovascular alterations induced by intermittent hypoxia. Front Physiol 5: 468, 2014. |
| 154. | Izal‐Azcarate A, Belzunegui S, San Sebastian W, Garrido‐Gil P, Vazquez‐Claverie M, Lopez B, Marcilla I, Luquin MA. Immunohistochemical characterization of the rat carotid body. Respir Physiol Neurobiol 161: 95‐99, 2008. |
| 155. | Jonz MG, Buck LT, Perry SF, Schwerte T, Zaccone G. Sensing and surviving hypoxia in vertebrates. Ann N Y Acad Sci 1365: 43‐58, 2016. |
| 156. | Kapadia V, Rabi Y, Oei JL. The Goldilocks principle. Oxygen in the delivery room: When is it too little, too much, and just right? Semin Fetal Neonatal Med 23: 347‐354, 2018. |
| 157. | Karetzky MS, Keighley JF, Mithoefer JC. The effect of oxygen administration on gas exchange and cardiopulmonary function in normal subjects. Respir Physiol 12: 361‐370, 1971. |
| 158. | Katz DM. Regulation of respiratory neuron development by neurotrophic and transcriptional signaling mechanisms. Respir Physiol Neurobiol 149: 99‐109, 2005. |
| 159. | Katz‐Salamon M, Eriksson M, Jonsson B. Development of peripheral chemoreceptor function in infants with chronic lung disease and initially lacking hyperoxic response. Arch Dis Child Fetal Neonatal Ed 75: F4‐F9, 1996. |
| 160. | Katz‐Salamon M, Jonsson B, Lagercrantz H. Blunted peripheral chemoreceptor response to hyperoxia in a group of infants with bronchopulmonary dysplasia. Pediatr Pulmonol 20: 101‐106, 1995. |
| 161. | Katz‐Salamon M, Lagercrantz H. Hypoxic ventilatory defence in very preterm infants: Attenuation after long term oxygen treatment. Arch Dis Child Fetal Neonatal Ed 70: F90‐F95, 1994. |
| 162. | Kavanagh JB, Pratt AE, Lewallen RM, Bavis RW. Plasticity in normoxic ventilation and apnea frequency in neonatal rats exposed to chronic hyperoxia. FASEB J 32, 2018. (Abstract). |
| 163. | Kayton A, Timoney P, Vargo L, Perez JA. A review of oxygen physiology and appropriate management of oxygen levels in premature neonates. Adv Neonatal Care 18: 98‐104, 2018. |
| 164. | Kazemi H, Hoop B. Glutamic acid and gamma‐aminobutyric acid neurotransmitters in central control of breathing. J Appl Physiol 70: 1‐7, 1991. |
| 165. | Keys A, Stapp JP, Violante A. Responses in size, output and efficiency of the human heart to acute alteration in the composition of inspired air. Am J Physiol 138: 763‐771, 1943. |
| 166. | Khazin AF, Hon EH, Hehre FW. Effects of maternal hyperoxia on the fetus. I. Oxygen tension. Am J Obstet Gynecol 109: 628‐637, 1971. |
| 167. | Kim D. K+ channels in O2 sensing and postnatal development of carotid body glomus cell response to hypoxia. Respir Physiol Neurobiol 185: 44‐56, 2013. |
| 168. | Kim D, Cavanaugh EJ, Kim I, Carroll JL. Heteromeric TASK‐1/TASK‐3 is the major oxygen‐sensitive background K+ channel in rat carotid body glomus cells. J Physiol 587: 2963‐2975, 2009. |
| 169. | Kim I, Donnelly DF, Carroll JL. Modulation of gene expression in subfamilies of TASK K+ channels by chronic hyperoxia exposure in rat carotid body. Adv Exp Med Biol 580: 37‐41; discussion 351‐359, 2006. |
| 170. | Kim I, Donnelly DF, Carroll JL. Postnatal hyperoxia impairs acute oxygen sensing of rat glomus cells by reduced membrane depolarization. Adv Exp Med Biol 758: 49‐54, 2012. |
| 171. | Kim I, Yang D, Carroll JL, Donnelly DF. Perinatal hyperoxia exposure impairs hypoxia‐induced depolarization in rat carotid body glomus cells. Respir Physiol Neurobiol 188: 9‐14, 2013. |
| 172. | Kinkead R, Gulemetova R. Neonatal maternal separation and neuroendocrine programming of the respiratory control system in rats. Biol Psychol 84: 26‐38, 2010. |
| 173. | Kline DD, Ogier M, Kunze DL, Katz DM. Exogenous brain‐derived neurotrophic factor rescues synaptic dysfunction in Mecp2‐null mice. J Neurosci 30: 5303‐5310, 2010. |
| 174. | Kron M, Lang M, Adams IT, Sceniak M, Longo F, Katz DM. A BDNF loop‐domain mimetic acutely reverses spontaneous apneas and respiratory abnormalities during behavioral arousal in a mouse model of Rett syndrome. Dis Model Mech 7: 1047‐1055, 2014. |
| 175. | Kumar P, Prabhakar NR. Peripheral chemoreceptors: Function and plasticity of the carotid body. Compr Physiol 2: 141‐219, 2012. |
| 176. | Lahiri S. Role of arterial O2 flow in peripheral chemoreceptor excitation. Fed Proc 39: 2648‐2652, 1980. |
| 177. | Lahiri S, Mokashi A, Shirahata M, Andronikou S. Chemical respiratory control in chronically hyperoxic cats. Respir Physiol 82: 201‐215, 1990. |
| 178. | Lahiri S, Mulligan E, Andronikou S, Shirahata M, Mokashi A. Carotid body chemosensory function in prolonged normobaric hyperoxia in the cat. J Appl Physiol 62: 1924‐1931, 1987. |
| 179. | Lambertsen CJ, Ewing JH, Kough RH, Gould R, Stroud MW III. Oxygen toxicity; arterial and internal jugular blood gas composition in man during inhalation of air, 100% O2 and 2% CO2 in O2 at 3.5 atmospheres ambient pressure. J Appl Physiol 8: 255‐263, 1955. |
| 180. | Lambertsen CJ, Stroud MW, Gould RA, Kough RH, Ewing JH, Schmidt CF. Oxygen toxicity; respiratory responses of normal men to inhalation of 6 and 100 per cent oxygen under 3.5 atmospheres pressure. J Appl Physiol 5: 487‐493, 1953. |
| 181. | Latzin P, Roth S, Thamrin C, Hutten GJ, Pramana I, Kuehni CE, Casaulta C, Nelle M, Riedel T, Frey U. Lung volume, breathing pattern and ventilation inhomogeneity in preterm and term infants. PLoS One 4: e4635, 2009. |
| 182. | Lau AG, Irier HA, Gu J, Tian D, Ku L, Liu G, Xia M, Fritsch B, Zheng JQ, Dingledine R, Xu B, Lu B, Feng Y. Distinct 3'UTRs differentially regulate activity‐dependent translation of brain‐derived neurotrophic factor (BDNF). Proc Natl Acad Sci U S A 107: 15945‐15950, 2010. |
| 183. | Leite MS, Damaceno‐Rodrigues NR, Simone MR, Santos AB, Bueno HM, Battlehner CN, Mauad T, Caldini EG, Saldiva PH. Structural alterations in adult rat carotid bodies exposed to hyperbaric oxygenation. Undersea Hyperb Med 37: 419‐432, 2010. |
| 184. | Lenfant C. Arterial‐alveolar difference in PCO2 during air and oxygen breathing. J Appl Physiol 21: 1356‐1362, 1966. |
| 185. | Leonard EM, Salman S, Nurse CA. Sensory processing and integration at the carotid body tripartite synapse: Neurotransmitter functions and effects of chronic hypoxia. Front Physiol 9: 225, 2018. |
| 186. | Liberzon I, Arieli R, Kerem D. Attenuation of hypoxic ventilation by hyperbaric O2: Effects of pressure and exposure time. J Appl Physiol 66: 851‐856, 1989. |
| 187. | Lim K, Wheeler KI, Gale TJ, Jackson HD, Kihlstrand JF, Sand C, Dawson JA, Dargaville PA. Oxygen saturation targeting in preterm infants receiving continuous positive airway pressure. J Pediatr 164: 730‐736. e731, 2014. |
| 188. | Lin TB, Lo MJ, Huang CY, Ting H, Lee SD. GABAergic modulation of ventilatory response to acute and sustained hypoxia in obese Zucker rats. Int J Obes (Lond) 29: 188‐195, 2005. |
| 189. | Ling L, Olson EB Jr, Vidruk EH, Mitchell GS. Attenuation of the hypoxic ventilatory response in adult rats following one month of perinatal hyperoxia. J Physiol 495 (Pt 2): 561‐571, 1996. |
| 190. | Ling L, Olson EB Jr, Vidruk EH, Mitchell GS. Integrated phrenic responses to carotid afferent stimulation in adult rats following perinatal hyperoxia. J Physiol 500 (Pt 3): 787‐796, 1997. |
| 191. | Ling L, Olson EB Jr, Vidruk EH, Mitchell GS. Phrenic responses to isocapnic hypoxia in adult rats following perinatal hyperoxia. Respir Physiol 109: 107‐116, 1997. |
| 192. | Ling L, Olson EB Jr, Vidruk EH, Mitchell GS. Slow recovery of impaired phrenic responses to hypoxia following perinatal hyperoxia in rats. J Physiol 511 (Pt 2): 599‐603, 1998. |
| 193. | Ling L, Olson EB Jr, Vidruk EH, Mitchell GS. Developmental plasticity of the hypoxic ventilatory response. Respir Physiol 110: 261‐268, 1997. |
| 194. | Liu Q, Lowry TF, Wong‐Riley MT. Postnatal changes in ventilation during normoxia and acute hypoxia in the rat: Implication for a sensitive period. J Physiol 577: 957‐970, 2006. |
| 195. | Lofaso F, Dauger S, Matrot B, Vardon G, Gaultier C, Gallego J. Inhibitory effects of repeated hyperoxia on breathing in newborn mice. Eur Respir J 29: 18‐24, 2007. |
| 196. | Logan S, Tobin KE, Fallon SC, Deng KS, McDonough AB, Bavis RW. Chronic intermittent hyperoxia alters the development of the hypoxic ventilatory response in neonatal rats. Respir Physiol Neurobiol 220: 69‐80, 2016. |
| 197. | Mankouski A, Kantores C, Wong MJ, Ivanovska J, Jain A, Benner EJ, Mason SN, Tanswell AK, Auten RL, Jankov RP. Intermittent hypoxia during recovery from neonatal hyperoxic lung injury causes long‐term impairment of alveolar development: A new rat model of BPD. Am J Physiol Lung Cell Mol Physiol 312: L208‐L216, 2017. |
| 198. | Marczak M, Pokorski M. Oxygen breathing and ventilation. J Physiol Pharmacol 55: 127‐134, 2004. |
| 199. | Martin‐Body RL, Robson GJ, Sinclair JD. Respiratory effects of sectioning the carotid sinus glossopharyngeal and abdominal vagal nerves in the awake rat. J Physiol 361: 35‐45, 1985. |
| 200. | Martin‐Body RL, Robson GJ, Sinclair JD. Restoration of hypoxic respiratory responses in the awake rat after carotid body denervation by sinus nerve section. J Physiol 380: 61‐73, 1986. |
| 201. | Massari VJ, Shirahata M, Johnson TA, Gatti PJ. Carotid sinus nerve terminals which are tyrosine hydroxylase immunoreactive are found in the commissural nucleus of the tractus solitarius. J Neurocytol 25: 197‐208, 1996. |
| 202. | Matalon S, Nesarajah MS, Farhi LE. Pulmonary and circulatory changes in conscious sheep exposed to 100% O2 at 1 ATA. J Appl Physiol 53: 110‐116, 1982. |
| 203. | Matalon SV, Manning PJ, Bernie BJ, Eichorst BC, Hunt CE, Seeds AE. The effects of changes of maternal PaO2 and PaCO2 on the fetal PaO2 and PaCO2—In vivo study. Respir Physiol 32: 51‐61, 1978. |
| 204. | Matott MP, Ciarlone GE, Putnam RW, Dean JB. Normobaric hyperoxia (95% O2) stimulates CO2‐sensitive and CO2‐insensitive neurons in the caudal solitary complex of rat medullary tissue slices maintained in 40% O2. Neuroscience 270: 98‐122, 2014. |
| 205. | May P. L′action immediate de l'oxygene sur la ventilation chez l'homme normal. Helv Physiol Pharmacol Acta 15: 230‐240, 1957. |
| 206. | Miller MJ, Tenney SM. Hyperoxic hyperventilation in carotid‐deafferented cats. Respir Physiol 23: 23‐30, 1975. |
| 207. | Milsom WK, Burleson ML. Peripheral arterial chemoreceptors and the evolution of the carotid body. Respir Physiol Neurobiol 157: 4‐11, 2007. |
| 208. | Mitchell GS, Johnson SM. Neuroplasticity in respiratory motor control. J Appl Physiol 94: 358‐374, 2003. |
| 209. | Mokashi A, Di Guilio C, Morelli L, Lahiri S. Chronic hyperoxic effects on cat carotid body catecholamines and structure. Respir Physiol 97: 25‐32, 1994. |
| 210. | Mokashi A, Lahiri S. Aortic and carotid body chemoreception in prolonged hyperoxia in the cat. Respir Physiol 86: 233‐243, 1991. |
| 211. | Montandon G, Kinkead R, Bairam A. Adenosinergic modulation of respiratory activity: Developmental plasticity induced by perinatal caffeine administration. Respir Physiol Neurobiol 164: 87‐95, 2008. |
| 212. | Moore PK, Handy RL. Selective inhibitors of neuronal nitric oxide synthase—Is no NOS really good NOS for the nervous system? Trends Pharmacol Sci 18: 204‐211, 1997. |
| 213. | Morgan BJ, Adrian R, Bates ML, Dopp JM, Dempsey JA. Quantifying hypoxia‐induced chemoreceptor sensitivity in the awake rodent. J Appl Physiol 117: 816‐824, 2014. |
| 214. | Mortola JP. Prenatal hyperoxia blunts the hypoxic ventilatory chemosensitivity of the 1‐day old chicken hatchling. Respir Physiol Neurobiol 178: 352‐356, 2011. |
| 215. | Mouradian GC Jr, Alvarez‐Argote S, Gorzek R, Thuku G, Michkalkiewicz T, Wong‐Riley MTT, Konduri GG, Hodges MR. Acute and chronic changes in the control of breathing in a rat model of bronchopulmonary dysplasia. Am J Physiol Lung Cell Mol Physiol 316: L506‐L518, 2019. |
| 216. | Mu L, Xia DD, Michalkiewicz T, Hodges M, Mouradian G, Konduri GG, Wong‐Riley MTT. Effects of neonatal hyperoxia on the critical period of postnatal development of neurochemical expressions in brain stem respiratory‐related nuclei in the rat. Physiol Rep 6, 2018. DOI: 10.14814/phy2.13627. |
| 217. | Mulkey DK, Henderson RA III, Olson JE, Putnam RW, Dean JB. Oxygen measurements in brain stem slices exposed to normobaric hyperoxia and hyperbaric oxygen. J Appl Physiol 90: 1887‐1899, 2001. |
| 218. | Mulkey DK, Henderson RA III, Putnam RW, Dean JB. Hyperbaric oxygen and chemical oxidants stimulate CO2/H+‐sensitive neurons in rat brain stem slices. J Appl Physiol 95: 910‐921, 2003. |
| 219. | Mulkey DK, Henderson RA III, Ritucci NA, Putnam RW, Dean JB. Oxidative stress decreases pHi and Na+/H+ exchange and increases excitability of solitary complex neurons from rat brain slices. Am J Physiol Cell Physiol 286: C940‐C951, 2004. |
| 220. | Muscato GM, Kim MS, McDonough AB, Bavis RW. Does reduced carotid body BDNF contribute to developmental hyperoxia‐induced respiratory plasticity? FASEB J 31, 2017. (Abstract). |
| 221. | Nardiello C, Mizikova I, Morty RE. Looking ahead: Where to next for animal models of bronchopulmonary dysplasia? Cell Tissue Res 367: 457‐468, 2017. |
| 222. | Ogawa H, Mizusawa A, Kikuchi Y, Hida W, Miki H, Shirato K. Nitric oxide as a retrograde messenger in the nucleus tractus solitarii of rats during hypoxia. J Physiol 486 (Pt 2): 495‐504, 1995. |
| 223. | Ogier M, Kron M, Katz DM. Neurotrophic factors in development and regulation of respiratory control. Compr Physiol 3: 1125‐1134, 2013. |
| 224. | Ohtake PJ, Simakajornboon N, Fehniger MD, Xue YD, Gozal D. N‐Methyl‐D‐aspartate receptor expression in the nucleus tractus solitarii and maturation of hypoxic ventilatory response in the rat. Am J Respir Crit Care Med 162: 1140‐1147, 2000. |
| 225. | Olson EB Jr, Vidruk EH, Dempsey JA. Carotid body excision significantly changes ventilatory control in awake rats. J Appl Physiol 64: 666‐671, 1988. |
| 226. | O'Reilly M, Thebaud B. Animal models of bronchopulmonary dysplasia. The term rat models. Am J Physiol Lung Cell Mol Physiol 307: L948‐L958, 2014. |
| 227. | Oswald MCW, Garnham N, Sweeney ST, Landgraf M. Regulation of neuronal development and function by ROS. FEBS Lett 592: 679‐691, 2018. |
| 228. | Otis AB, Rahn H, Brontman M, Mullins LJ, Fenn WO. Ballistocardiographic study of changes in cardiac output due to respiration. J Clin Investig 25: 413‐421, 1946. |
| 229. | Paton JF. A working heart‐brainstem preparation of the mouse. J Neurosci Methods 65: 63‐68, 1996. |
| 230. | Pawar A, Nanduri J, Yuan G, Khan SA, Wang N, Kumar GK, Prabhakar NR. Reactive oxygen species‐dependent endothelin signaling is required for augmented hypoxic sensory response of the neonatal carotid body by intermittent hypoxia. Am J Physiol 296: R735‐R742, 2009. |
| 231. | Pawar A, Peng YJ, Jacono FJ, Prabhakar NR. Comparative analysis of neonatal and adult rat carotid body responses to chronic intermittent hypoxia. J Appl Physiol 104: 1287‐1294, 2008. |
| 232. | Peng YJ, Rennison J, Prabhakar NR. Intermittent hypoxia augments carotid body and ventilatory response to hypoxia in neonatal rat pups. J Appl Physiol 97: 2020‐2025, 2004. |
| 233. | Penn JS, Henry MM, Wall PT, Tolman BL. The range of PaO2 variation determines the severity of oxygen‐induced retinopathy in newborn rats. Invest Ophthalmol Vis Sci 36: 2063‐2070, 1995. |
| 234. | Perry SF, Tzaneva V. The sensing of respiratory gases in fish: Mechanisms and signalling pathways. Respir Physiol Neurobiol 224: 71‐79, 2016. |
| 235. | Pilla R, Landon CS, Dean JB. A potential early physiological marker for CNS oxygen toxicity: Hyperoxic hyperpnea precedes seizure in unanesthetized rats breathing hyperbaric oxygen. J Appl Physiol 114: 1009‐1020, 2013. |
| 236. | Piskuric NA, Nurse CA. Expanding role of ATP as a versatile messenger at carotid and aortic body chemoreceptors. J Physiol 591: 415‐422, 2013. |
| 237. | Piskuric NA, Zhang M, Vollmer C, Nurse CA. Potential roles of ATP and local neurons in the monitoring of blood O2 content by rat aortic bodies. Exp Physiol 99: 248‐261, 2014. |
| 238. | Poff AM, Kernagis D, D'Agostino DP. Hyperbaric environment: Oxygen and cellular damage versus protection. Compr Physiol 7: 213‐234, 2016. |
| 239. | Pokorski M, Kolesnikova E, Marczak M, Budzinska K. Neurotransmitter mechanisms in the enhancement of the hypoxic ventilatory response by antecedent hyperoxia in the anesthetized rat. J Physiol Pharmacol 56: 433‐446, 2005. |
| 240. | Pokorski M, Marczak M, Jernajczyk U. Augmentation of hypoxic respiration after brief hyperoxia in the anesthetized cat: Putative function of GABAA neurotransmission. J Biomed Sci 11: 322‐330, 2004. |
| 241. | Porzionato A, Macchi V, Parenti A, De Caro R. Trophic factors in the carotid body. Int Rev Cell Mol Biol 269: 1‐58, 2008. |
| 242. | Prabhakar NR, Peng YJ, Kumar GK, Nanduri J. Peripheral chemoreception and arterial pressure responses to intermittent hypoxia. Compr Physiol 5: 561‐577, 2015. |
| 243. | Prabhakar NR, Peng YJ, Kumar GK, Pawar A. Altered carotid body function by intermittent hypoxia in neonates and adults: Relevance to recurrent apneas. Respir Physiol Neurobiol 157: 148‐153, 2007. |
| 244. | Prabhakar NR, Peng YJ, Yuan G, Nanduri J. Reactive oxygen radicals and gaseous transmitters in carotid body activation by intermittent hypoxia. Cell Tissue Res 372: 427‐431, 2018. |
| 245. | Pratt AE, Bavis RW. Respiratory plasticity in adult rats after exposure to chronic hyperoxia. FASEB J 33, 2019. (Abstract). |
| 246. | Prieto‐Lloret J, Caceres AI, Obeso A, Rocher A, Rigual R, Agapito MT, Bustamante R, Castaneda J, Perez‐Garcia MT, Lopez‐Lopez JR, Gonzalez C. Ventilatory responses and carotid body function in adult rats perinatally exposed to hyperoxia. J Physiol 554: 126‐144, 2004. |
| 247. | Prieto‐Lloret J, Ramirez M, Olea E, Moral‐Sanz J, Cogolludo A, Castaneda J, Yubero S, Agapito T, Gomez‐Nino A, Rocher A, Rigual R, Obeso A, Perez‐Vizcaino F, Gonzalez C. Hypoxic pulmonary vasoconstriction, carotid body function and erythropoietin production in adult rats perinatally exposed to hyperoxia. J Physiol 593: 2459‐2477, 2015. |
| 248. | Rakoczy RJ, Wyatt CN. Acute oxygen sensing by the carotid body: A rattlebag of molecular mechanisms. J Physiol 596: 2969‐2976, 2018. |
| 249. | Ramirez SC, Koschnitzky JE, Youngquist TM, Baertsch NA, Smith CV, Ramirez JM. Perinatal breathing patterns and survival in mice born prematurely and at term. Front Physiol 10: 1113, 2019. |
| 250. | Reiner A, Zagvazdin Y. On the selectivity of 7‐nitroindazole as an inhibitor of neuronal nitric oxide synthase. Trends Pharmacol Sci 19: 348‐350, 1998. |
| 251. | Reinstorff D, Fenner A. Ventilatory response to hyperoxia in premature and newborn infants during the first three days of life. Respir Physiol 15: 159‐165, 1972. |
| 252. | Ren X, Fatemian M, Robbins PA. Changes in respiratory control in humans induced by 8 h of hyperoxia. J Appl Physiol 89: 655‐662, 2000. |
| 253. | Rigatto H, Brady JP, de la Torre Verduzco R. Chemoreceptor reflexes in preterm infants: I. The effect of gestational and postnatal age on the ventilatory response to inhalation of 100% and 15% oxygen. Pediatrics 55: 604‐613, 1975. |
| 254. | Roeser JC, Brackett DG, van Heerden ES, Young KM, Bavis RW. Potentiation of the hypoxic ventilatory response by 1 day of hyperoxia in neonatal rats. Respir Physiol Neurobiol 176: 50‐56, 2011. |
| 255. | Sah R, Galeffi F, Ahrens R, Jordan G, Schwartz‐Bloom RD. Modulation of the GABAA‐gated chloride channel by reactive oxygen species. J Neurochem 80: 383‐391, 2002. |
| 256. | Sah R, Schwartz‐Bloom RD. Optical imaging reveals elevated intracellular chloride in hippocampal pyramidal neurons after oxidative stress. J Neurosci 19: 9209‐9217, 1999. |
| 257. | Sankaran K, Wiebe H, Seshia MM, Boychuk RB, Cates D, Rigatto H. Immediate and late ventillatory response to high and low O2 in preterm infants and adult subjects. Pediatr Res 13: 875‐878, 1979. |
| 258. | Saugstad OD, Oei JL, Lakshminrusimha S, Vento M. Oxygen therapy of the newborn from molecular understanding to clinical practice. Pediatr Res 85: 20‐29, 2019. |
| 259. | Schachat SR, Labandeira CC, Saltzman MR, Cramer BD, Payne JL, Boyce CK. Phanerozoic pO2 and the early evolution of terrestrial animals. Proc Biol Sci 285: 20172631, 2018. |
| 260. | Serra A, Brozoski D, Hedin N, Franciosi R, Forster HV. Mortality after carotid body denervation in rats. J Appl Physiol 91: 1298‐1306, 2001. |
| 261. | Serra A, Brozoski D, Simeon T, Yi J, Bastasic J, Franciosi R, Jacobs ER, Forster HV. Serotonin and serotonin receptor expression in the aorta of carotid intact and denervated newborns. Respir Physiol Neurobiol 132: 253‐264, 2002. |
| 262. | Shephard RJ. Respiratory responses to the inhalation of oxygen at atmospheric pressure in normal subjects and in cases of congenital heart disease. J Physiol 127: 498‐514, 1955. |
| 263. | Shock NW, Soley MH. Effect of breathing pure oxygen on respiratory volume in humans. Proc Soc Exp Biol Med 44: 418‐420, 1940. |
| 264. | Simakajornboon N, Kuptanon T. Maturational changes in neuromodulation of central pathways underlying hypoxic ventilatory response. Respir Physiol Neurobiol 149: 273‐286, 2005. |
| 265. | Sladek M, Parker RA, Grogaard JB, Sundell HW. Long‐lasting effect of prolonged hypoxemia after birth on the immediate ventilatory response to changes in arterial partial pressure of oxygen in young lambs. Pediatr Res 34: 821‐828, 1993. |
| 266. | Smith CA, Forster HV, Blain GM, Dempsey JA. An interdependent model of central/peripheral chemoreception: Evidence and implications for ventilatory control. Respir Physiol Neurobiol 173: 288‐297, 2010. |
| 267. | Spicer JI, Burggren WW. Development of physiological regulatory systems: Altering the timing of crucial events. Zoology 106: 91‐99, 2003. |
| 268. | Stenson B, Saugstad OD. Oxygen treatment for immature infants beyond the delivery room: Lessons from randomized studies. J Pediatr 200: 12‐18, 2018. |
| 269. | Sterni LM, Bamford OS, Wasicko MJ, Carroll JL. Chronic hypoxia abolished the postnatal increase in carotid body type I cell sensitivity to hypoxia. Am J Physiol 277: L645‐L652, 1999. |
| 270. | Teppema LJ, Dahan A. The ventilatory response to hypoxia in mammals: Mechanisms, measurement, and analysis. Physiol Rev 90: 675‐754, 2010. |
| 271. | Thomson L, Paton J. Oxygen toxicity. Paediatr Respir Rev 15: 120‐123, 2014. |
| 272. | Torbati D, Mokashi A, Lahiri S. Effects of acute hyperbaric oxygenation on respiratory control in cats. J Appl Physiol 67: 2351‐2356, 1989. |
| 273. | Torbati D, Sherpa AK, Lahiri S, Mokashi A, Albertine KH, DiGiulio C. Hyperbaric oxygenation alters carotid body ultrastructure and function. Respir Physiol 92: 183‐196, 1993. |
| 274. | Torres JE, Kreisman NR, Gozal D. Nitric oxide modulates in vitro intrinsic optical signal and neural activity in the nucleus tractus solitarius of the rat. Neurosci Lett 232: 175‐178, 1997. |
| 275. | Torres‐Cuevas I, Parra‐Llorca A, Sanchez‐Illana A, Nunez‐Ramiro A, Kuligowski J, Chafer‐Pericas C, Cernada M, Escobar J, Vento M. Oxygen and oxidative stress in the perinatal period. Redox Biol 12: 674‐681, 2017. |
| 276. | Towell ME, Johnson J, Smedstad K, Andrew M, Vu TL. Fetal blood and tissue PO2 during maternal oxygen breathing. J Dev Physiol 6: 177‐185, 1984. |
| 277. | Truchot JP, Duhamel‐Jouve A. Oxygen and carbon dioxide in the marine intertidal environment: Diurnal and tidal changes in rockpools. Respir Physiol 39: 241‐254, 1980. |
| 278. | Vlasic V, Simakajornboon N, Gozal E, Gozal D. PDGF‐β receptor expression in the dorsocaudal brainstem parallels hypoxic ventilatory depression in the developing rat. Pediatr Res 50: 236‐241, 2001. |
| 279. | Vogel ER, Britt RD Jr, Trinidad MC, Faksh A, Martin RJ, MacFarlane PM, Pabelick CM, Prakash YS. Perinatal oxygen in the developing lung. Can J Physiol Pharmacol 93: 119‐127, 2015. |
| 280. | Vulesevic B, McNeill B, Perry SF. Chemoreceptor plasticity and respiratory acclimation in the zebrafish Danio rerio. J Exp Biol 209: 1261‐1273, 2006. |
| 281. | Vulesevic B, Perry SF. Developmental plasticity of ventilatory control in zebrafish, Danio rerio. Respir Physiol Neurobiol 154: 396‐405, 2006. |
| 282. | Waisman D, Arieli R, Kerem D, Melamed Y. Recovery of the hypoxic ventilatory drive of rats from the toxic effect of hyperbaric oxygen. Aviat Space Environ Med 63: 280‐286, 1992. |
| 283. | Wang H, Jafri A, Martin RJ, Nnanabu J, Farver C, Prakash YS, MacFarlane PM. Severity of neonatal hyperoxia determines structural and functional changes in developing mouse airway. Am J Physiol Lung Cell Mol Physiol 307: L295‐L301, 2014. |
| 284. | Wang J, Kim D. Activation of voltage‐dependent K+ channels strongly limits hypoxia‐induced elevation of [Ca2+]i in rat carotid body glomus cells. J Physiol 596: 3119‐3136, 2018. |
| 285. | Wang ZY, Bisgard GE. Postnatal growth of the carotid body. Respir Physiol Neurobiol 149: 181‐190, 2005. |
| 286. | Warner BB, Stuart LA, Papes RA, Wispe JR. Functional and pathological effects of prolonged hyperoxia in neonatal mice. Am J Physiol 275: L110‐L117, 1998. |
| 287. | Watt JG, Dumke PR, Comroe JHJ. Effects of inhalation of 100 per cent and 14 per cent oxygen upon respiration of unanesthetized dogs before and after chemoreceptor denervation. Am J Physiol 138: 610‐617, 1943. |
| 288. | Wenninger JM, Olson EB, Wang Z, Keith IM, Mitchell GS, Bisgard GE. Carotid sinus nerve responses and ventilatory acclimatization to hypoxia in adult rats following 2 weeks of postnatal hyperoxia. Respir Physiol Neurobiol 150: 155‐164, 2006. |
| 289. | Wilson RJ, Remmers JE, Paton JF. Brain stem PO2 and pH of the working heart‐brain stem preparation during vascular perfusion with aqueous medium. Am J Physiol Regul Integr Comp Physiol 281: R528‐R538, 2001. |
| 290. | Winn HR, Rubio R, Berne RM. Brain adenosine concentration during hypoxia in rats. Am J Physiol 241: H235‐H242, 1981. |
| 291. | Wong‐Riley MT, Liu Q. Neurochemical development of brain stem nuclei involved in the control of respiration. Respir Physiol Neurobiol 149: 83‐98, 2005. |
| 292. | Wong‐Riley MT, Liu Q. Neurochemical and physiological correlates of a critical period of respiratory development in the rat. Respir Physiol Neurobiol 164: 28‐37, 2008. |
| 293. | Wong‐Riley MTT, Liu Q, Gao X. Mechanisms underlying a critical period of respiratory development in the rat. Respir Physiol Neurobiol 264: 40‐50, 2019. |
| 294. | Wood JD, Watson WJ, Murray GW. Correlation between decreases in brain gamma‐aminobutyric acid levels and susceptibility to convulsions induced by hyperbaric oxygen. J Neurochem 16: 281‐287, 1969. |
| 295. | Yan S, Laferriere A, Zhang C, Moss IR. Microdialyzed adenosine in nucleus tractus solitarii and ventilatory response to hypoxia in piglets. J Appl Physiol 79: 405‐410, 1995. |
| 296. | Yang AL, Lo MJ, Ting H, Chen JS, Huang CY, Lee SD. GABAA and GABAB receptors differentially modulate volume and frequency in ventilatory compensation in obese Zucker rats. J Appl Physiol 102: 350‐357, 2007. |
| 297. | Yee M, Chess PR, McGrath‐Morrow SA, Wang Z, Gelein R, Zhou R, Dean DA, Notter RH, O'Reilly MA. Neonatal oxygen adversely affects lung function in adult mice without altering surfactant composition or activity. Am J Physiol Lung Cell Mol Physiol 297: L641‐L649, 2009. |
| 298. | Zapata P, Larrain C. How the carotid body works: Different strategies and preparations to solve different problems. Biol Res 38: 315‐328, 2005. |
| 299. | Zapata P, Stensaas LJ, Eyzaguirre C. Axon regeneration following a lesion of the carotid nerve: Electrophysiological and ultrastructural observations. Brain Res 113: 235‐253, 1976. |